
Bayesian inference of protein conformational ensembles from limited structural data
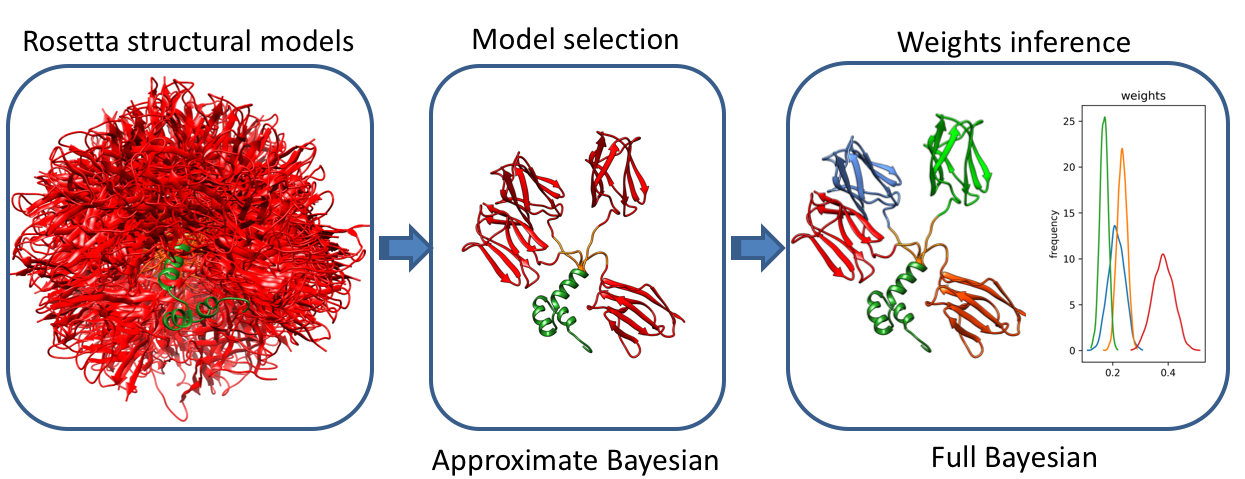
Many proteins consist of folded domains connected by regions with higher flexibility. The details of the resulting conformational ensemble play a central role in controlling interactions between domains and with binding partners. Small-Angle Scattering (SAS) is well-suited to study the conformational states adopted by proteins in solution. However, analysis is complicated by the limited information content in SAS data and care must be taken to avoid constructing overly complex ensemble models and fitting to noise in the experimental data. To address these challenges, we developed a method based on Bayesian statistics that infers conformational ensembles from a structural library generated by all-atom Monte Carlo simulations. The first stage of the method involves a fast model selection based on variational Bayesian inference that maximizes the model evidence of the selected ensemble. This is followed by a complete Bayesian inference of population weights in the selected ensemble. Experiments with simulated ensembles demonstrate that model evidence is capable of identifying the correct ensemble and that correct number of ensemble members can be recovered up to high level of noise. Using experimental data, we demonstrate how the method can be extended to include data from Nuclear Magnetic Resonance (NMR) and structural energies of conformers extracted from the all-atom energy functions. We show that the data from SAXS, NMR chemical shifts and energies calculated from conformers can work synergistically to improve the definition of the conformational ensemble.